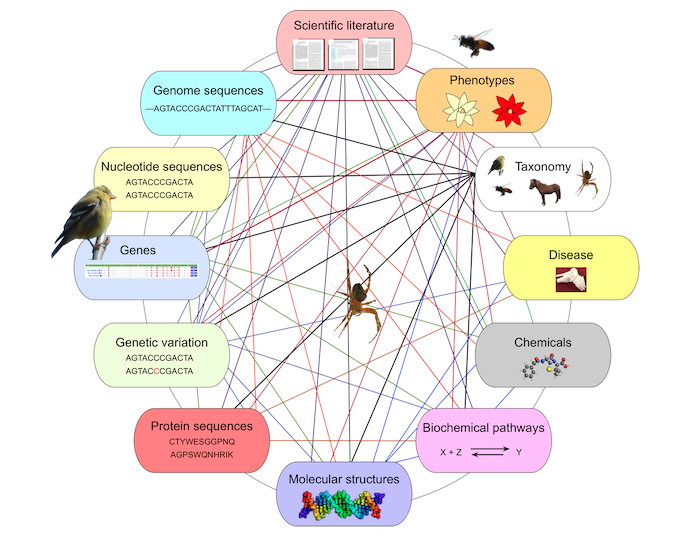
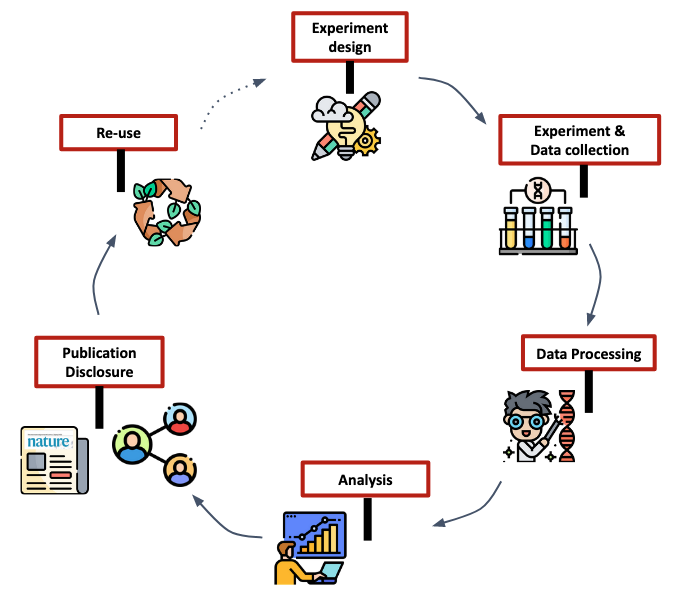
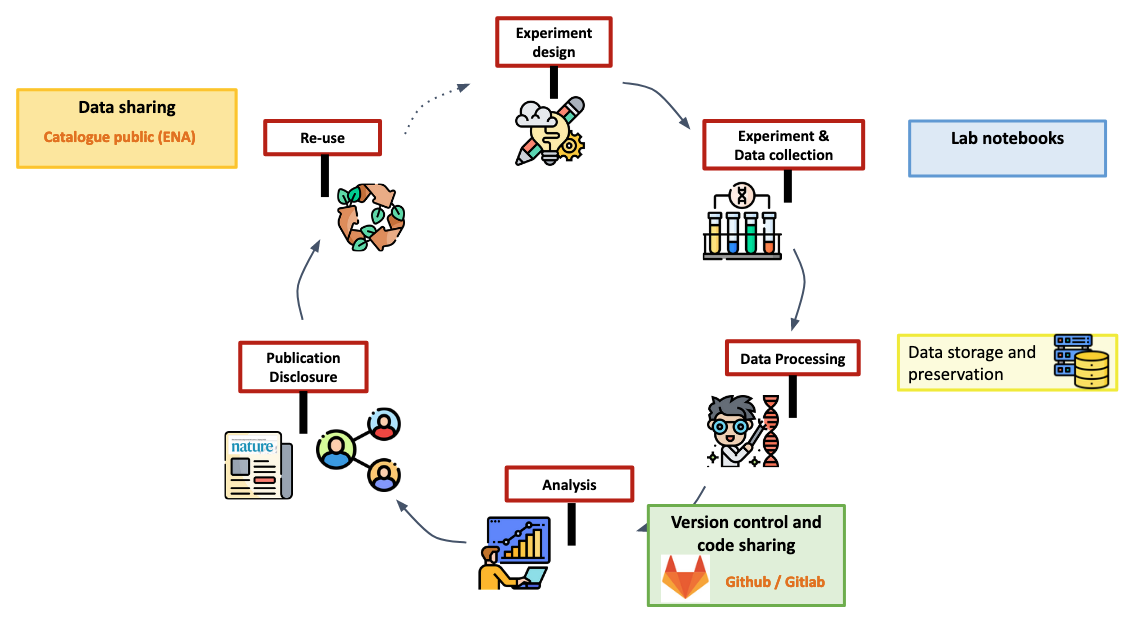
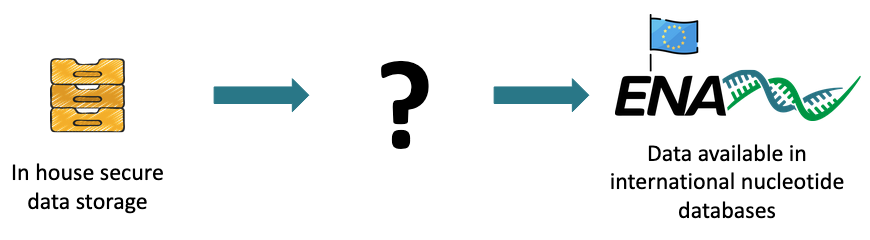Definition of biological databases
- Collection of structured biological data that can be accessed, searched and analyzed
- They play a crucial role in bioinformatics and computational biology : scientists can access to a wide variety of biological relevant data
- Organized in a collection of biological information : DNA, proteins sequences, molecular structure, gene expression data …
- Designed to facilitate efficient data storage, retrieval and analysis, allowing researchers to explore and interpret biological data on a large scale
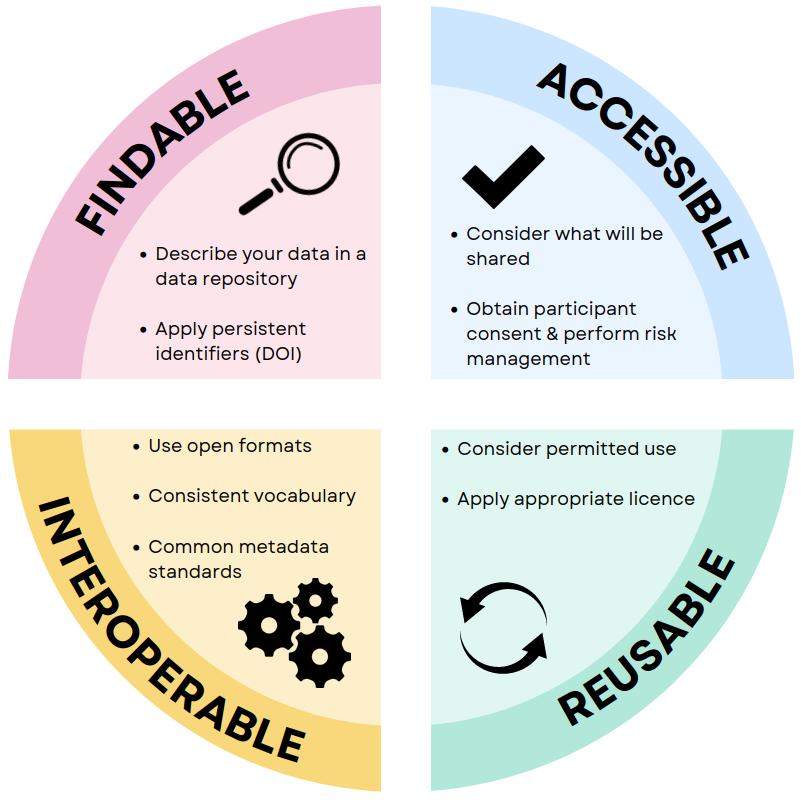
=> FAIR (Findable Accessible Interoperable Reusable) context!
![]()
image from https://digitalworldbiology.com/sites/default/files/DWB%20Product/teaser-images/exploring_databases.png
Types of biological databases : generalists or specialists
Types of biological databases : generalists or specialists
Specialists : databases that specialize in a particular organism or a particular type of data.
Classification of biological databases : Primary and secondary
Primary biological databases : data that it derives experimentally
Data : original and curated data
Raw experimental data
Purposes : data deposition and curation
Sources : researchers and laboratories
Original formats (fasta, fastq …)
Frequent updates and additions
Examples: GenBank (NCBI), EMBL-EBI, DDBJ (DNA Data Bank of Japan)
Classification of biological databases : Primary and secondary
Secondary biological databases : data obtained by analysis or treatment of data present in primary database
Data : derived data
Processed and annotated data
Purposes : data integration and analysis
Sources : primary databases
Standardized formats (GFF, BED …)
Updates based on primary databases
Examples : KEGG (Kyoto Encyclopedia of Genes and Genomics), Pfam (Protein Families), SCOP (Structural Classification Of Proteins)
Key properties of a well-designed biological databases
- Comprehensive
- Accuracy of data (quality control)
- Accessibility (user friendly interface)
- Interoperability (compatible with other databases or bioinformatics tools)
- Standardization (standards and formats for data representation, such as sequence file formats, ontologies, controlled vocabulary)
- Documentation (clear and metadata describing the data sources, data collection methods, and data processing procedures)
- Data security and privacy (sensitive nature)
- Updates and maintenance (new data, fix errors)
- Community engagement (encouraging user feed-back, collaboration and contribution)
- Long-term sustainability (supported by an organization or consortium)
=> FAIR PRINCIPLES!
Applications of biological databases : examples
- Genome annotation and analysis
- Protein structure prediction and modeling
- Genetic variations and disease-causing mutations
- Comparative genomics and evolutionary studies (Phylogeny, taxonomy)
- Gene expression analysis and functional genomics
- Drug discovery and target identification
- Systems biology and pathway analysis
=> Databases are essential tools for managing and utilizing data effectively in various fields and applications
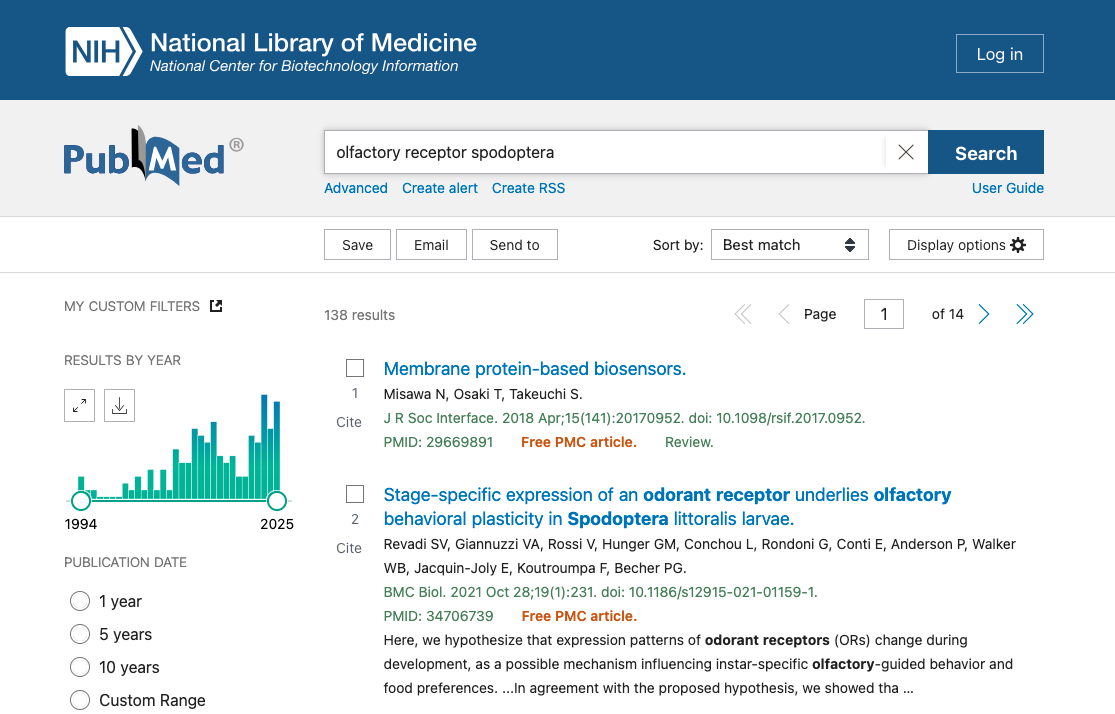
Main uses of biological databases
- Search data
- Download data
- Data analysis using integrated tools
- Submit data
Main uses of biological databases
Search data
Example : EBI
![]()
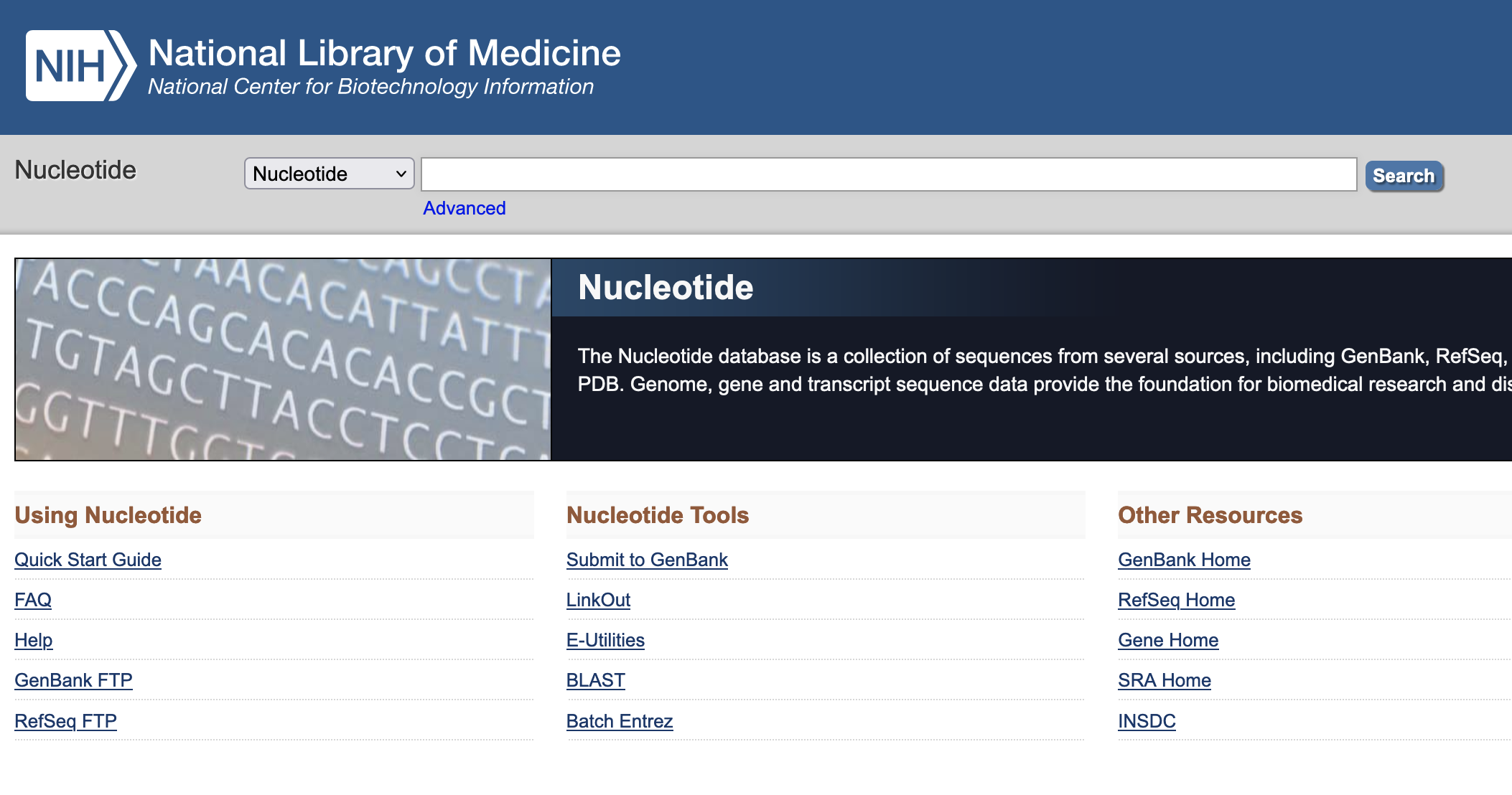
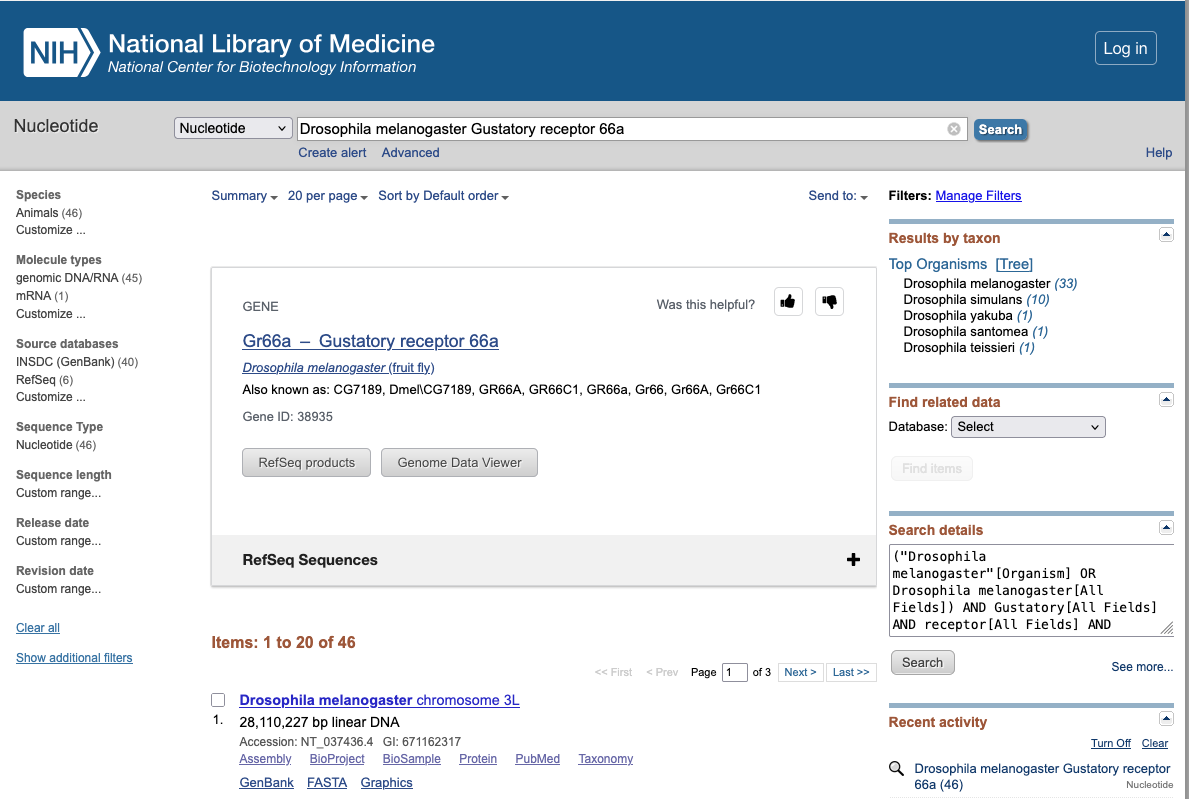
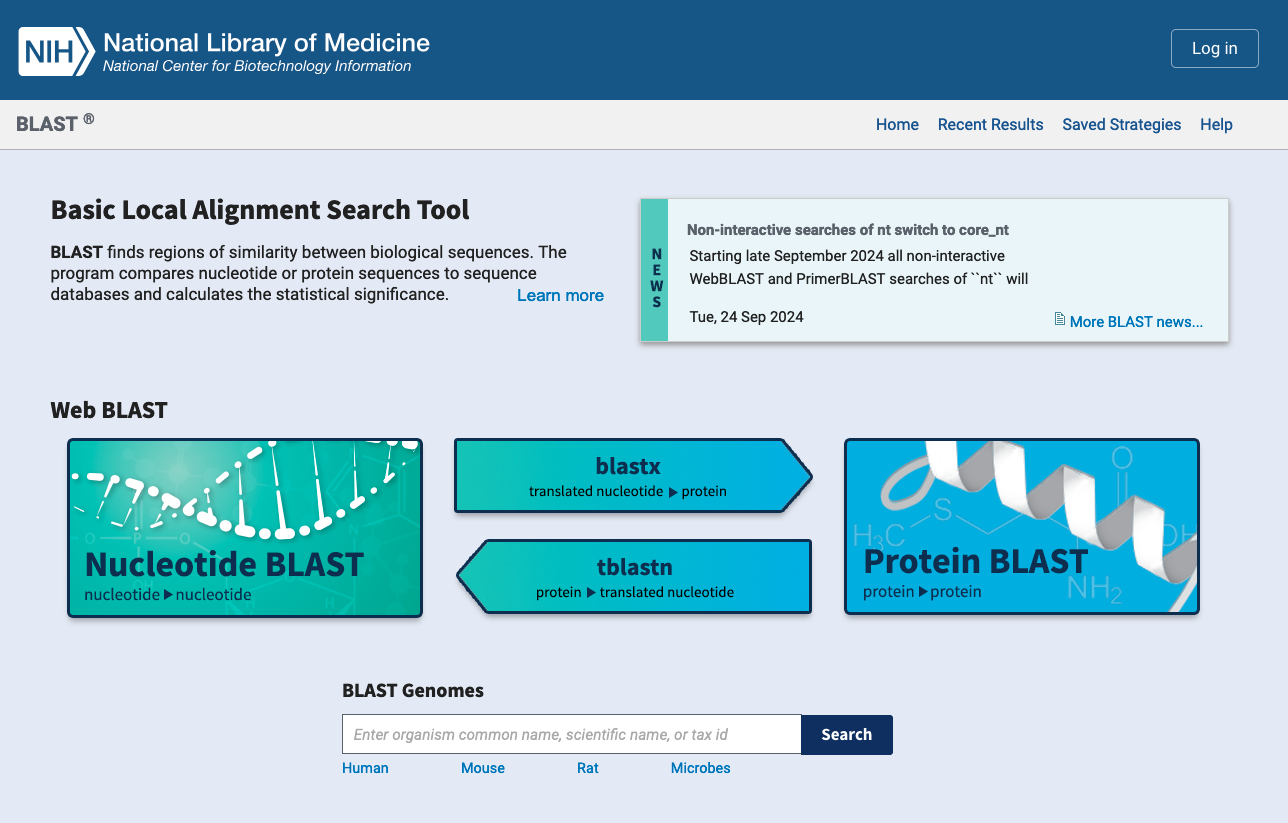
Main uses of biological databases
Download data
Example : NCBI
![]()
Main uses of biological databases
Example : NCBI Genome Data Viewer ![]()
Main uses of biological databases
Submit data
Example : EMBL-EBI submission ![]()
EMBL-EBI
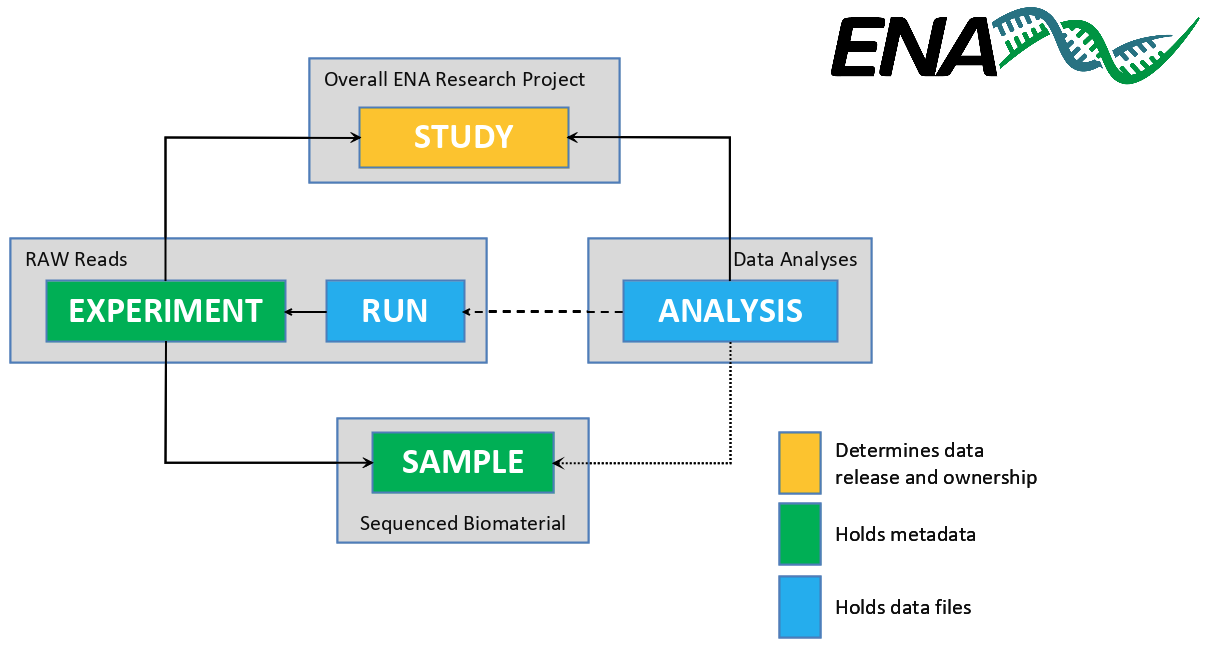
Example of DNA & RNA database : ENA (European Nucleotide Archive)
ENA (European Nucleotide Archive) : nucleotide sequencing information, covering raw sequencing data, sequence assembly information and functional annotation
![]()
EMBL-EBI
Example of DNA & RNA database : ENA (European Nucleotide Archive)
ENA (European Nucleotide Archive) : nucleotide sequencing information, covering raw sequencing data, sequence assembly information and functional annotation
![]()
EMBL-EBI
Example of DNA & RNA database : Ensembl
Ensembl : genome browser for animal genomes that supports research in comparative genomics, evolution, sequence variation and transcriptional regulation.
Ensembl annotates genes, computes multiple alignments, predicts regulatory function and collects disease data.
Ensembl tools include BLAST, BLAT, BioMart and the Variant Effect Predictor (VEP) for all supported species.
Release 115 (09/2025) : 348 species !
![]()
EMBL-EBI
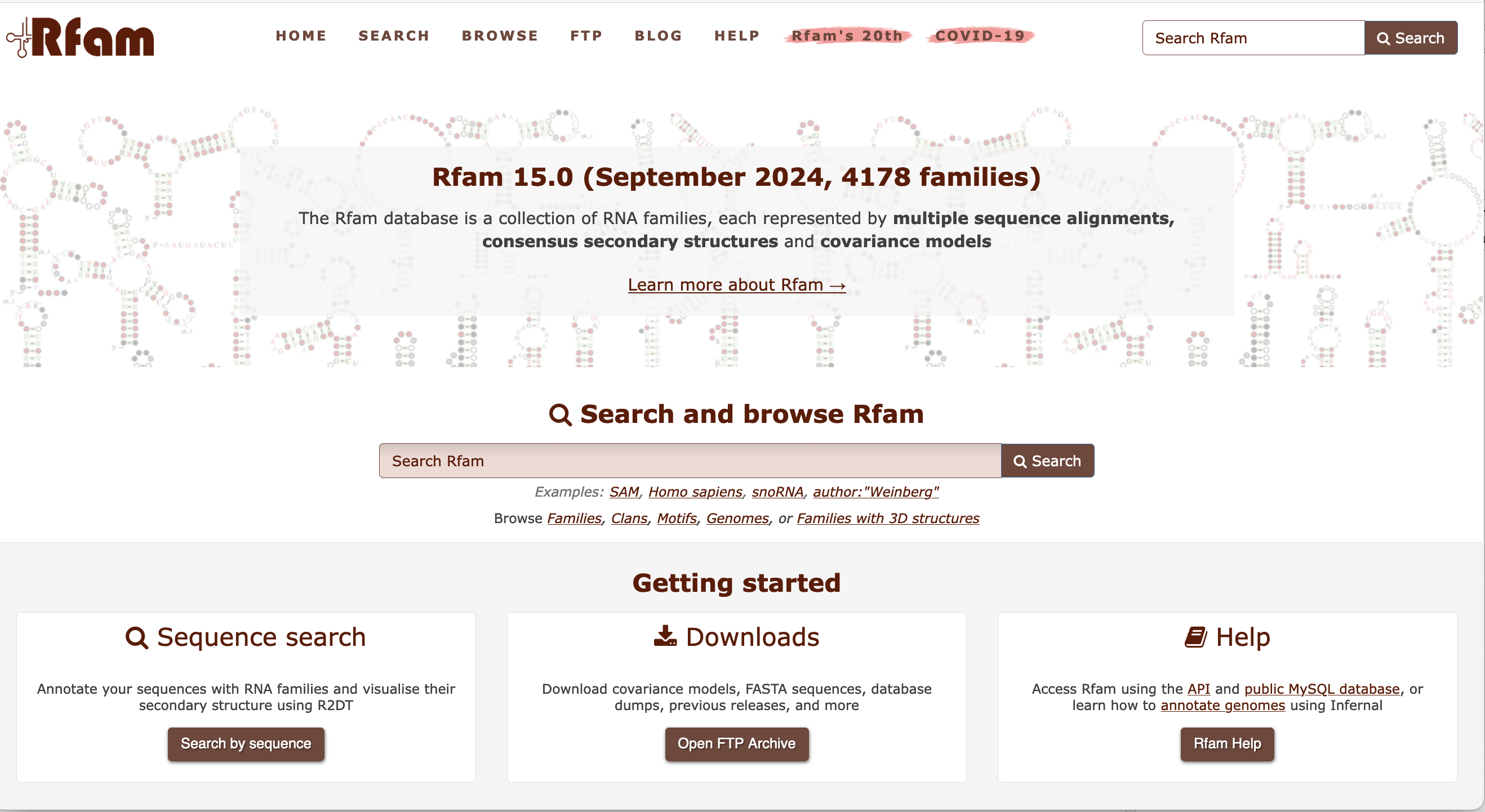
Example of DNA & RNA database : Rfam
Rfam : collection of RNA families, each represented by multiple sequence alignments, consensus secondary structures and covariance models
![]()
EMBL-EBI
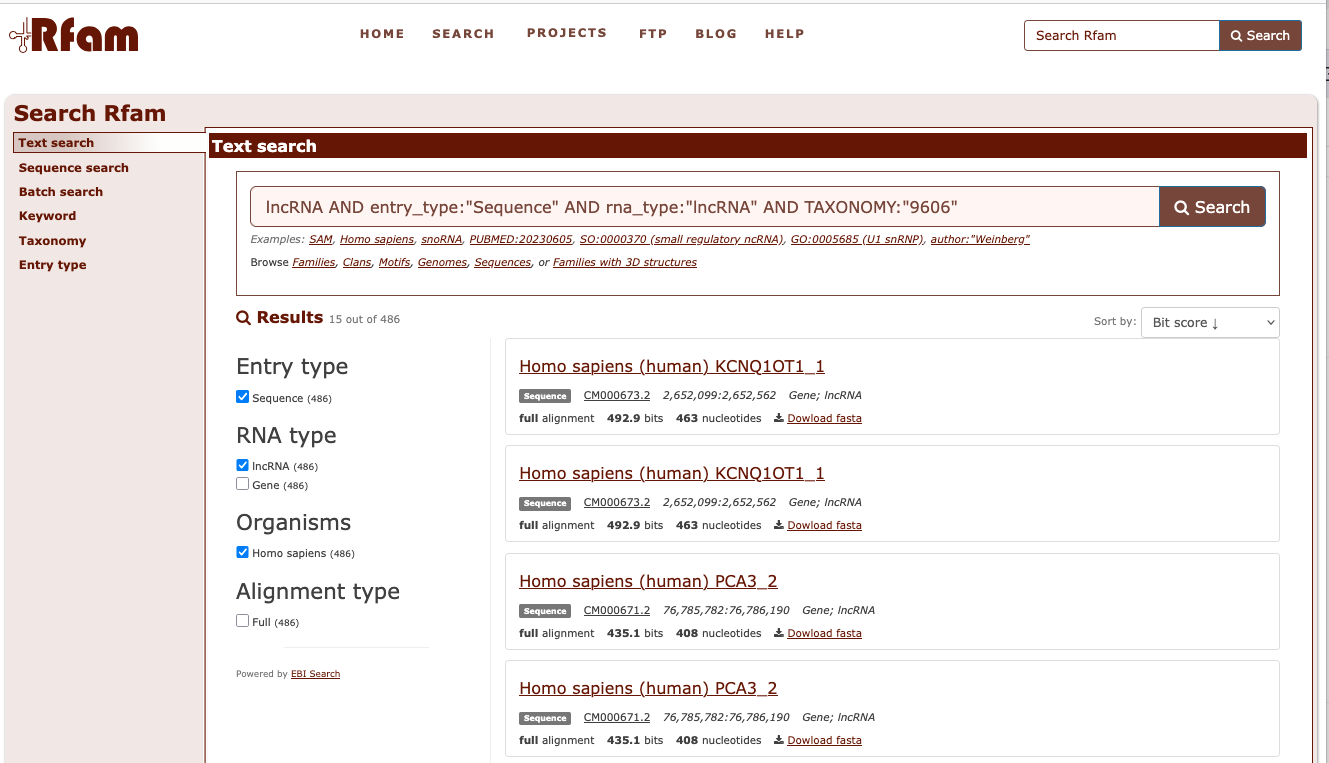
Example of DNA & RNA database : Rfam
Rfam
![]()
EMBL-EBI
Example of protein database : Uniprot
Uniprot : high-quality, comprehensive and freely accessible resource of protein sequence and functional information.
![]()
EMBL-EBI
Example of protein database : Uniprot
Uniprot
![]()
EMBL-EBI
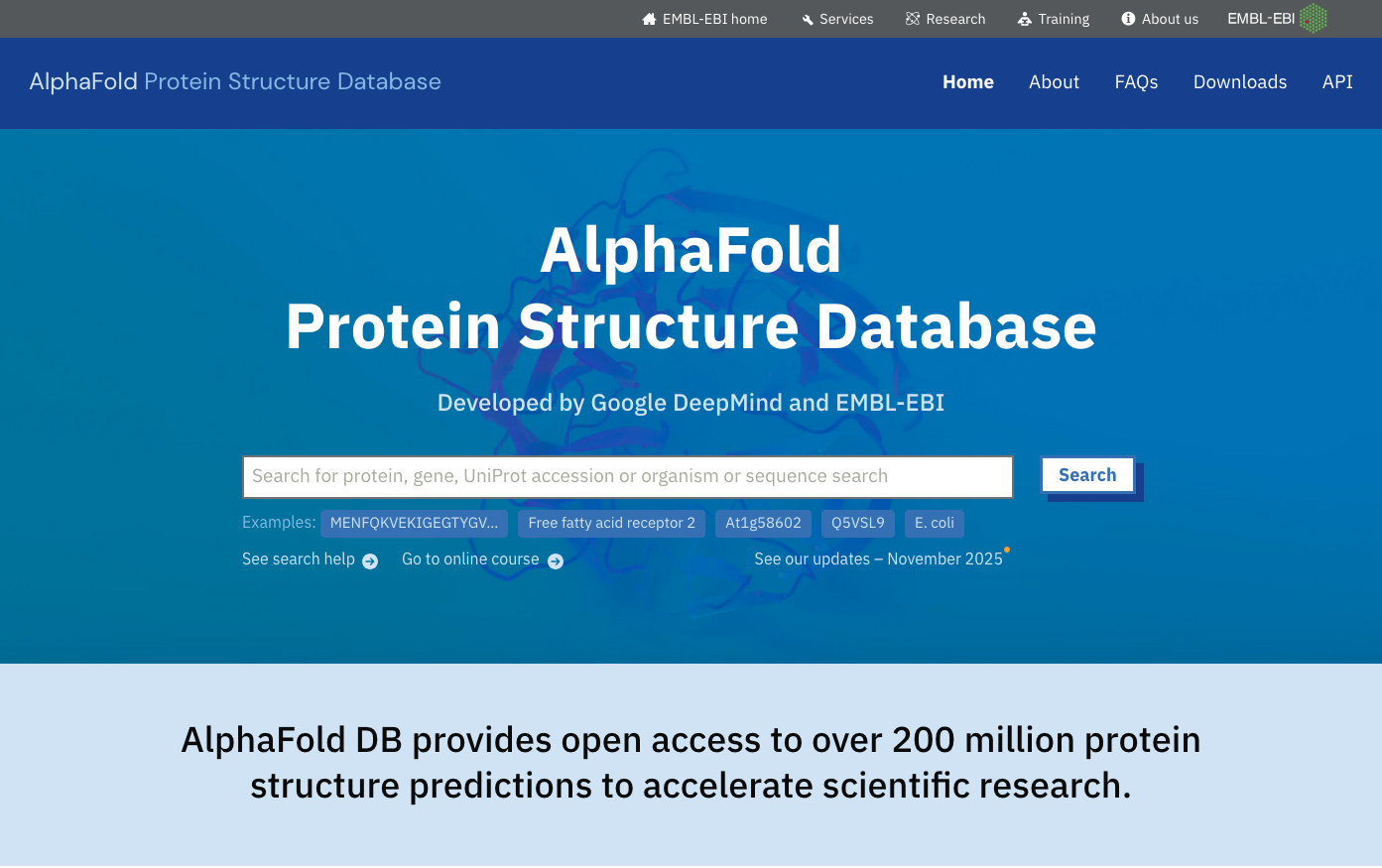
Example of protein database : AlphaFold
AlphaFold : database for protein structure predictions for numerous species
![]()
EMBL-EBI
Example of protein database : AlphaFold
AlphaFold
![]()
EMBL-EBI
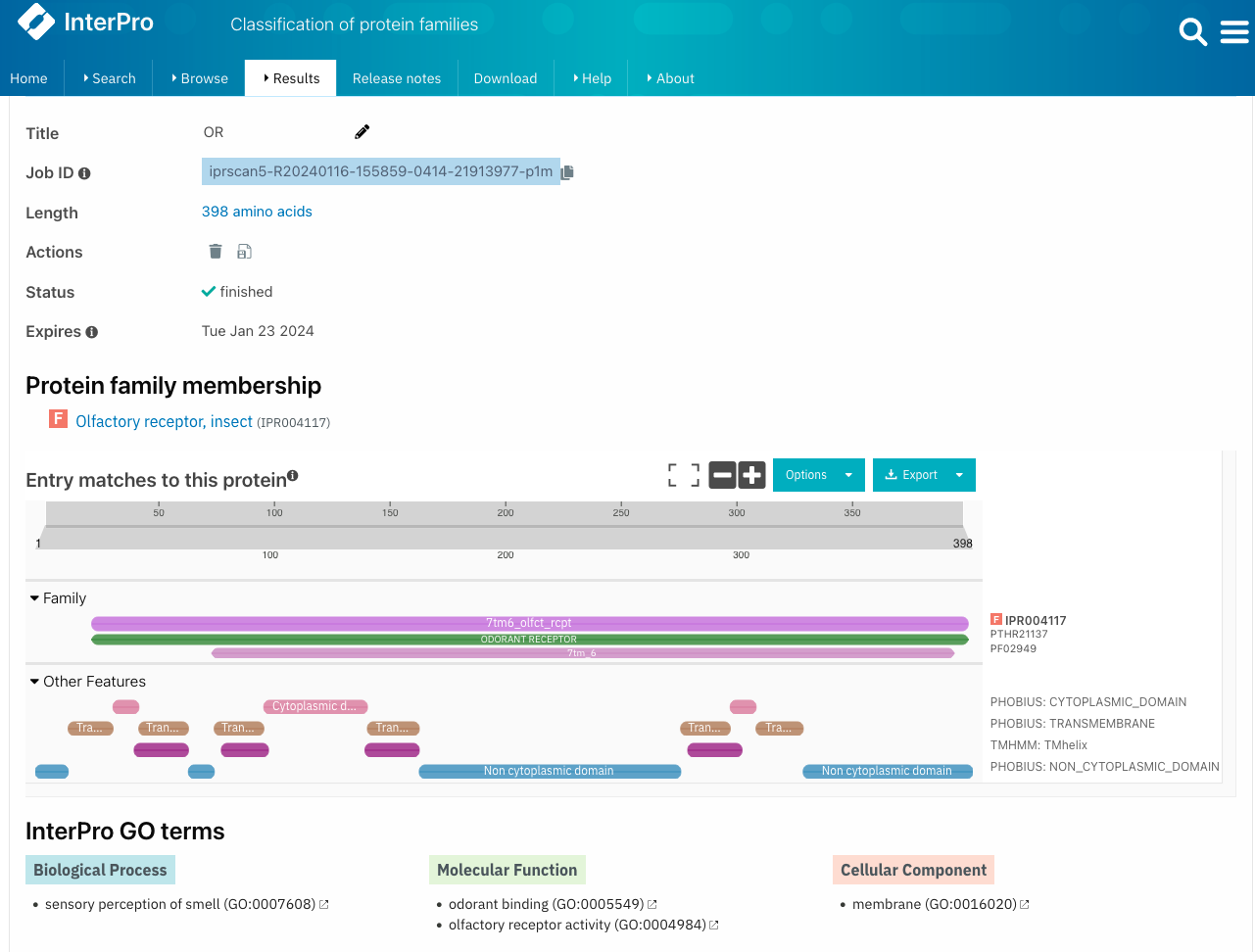
Example of protein database : InterProscan
InterProscan : functional analysis of proteins by classifying them into families and predicting domains and important sites
![]()
EMBL-EBI
Example of protein database : InterProscan
InterProscan
![]()
EMBL-EBI
Example of protein database : Pfam
Pfam : A database of conserved protein families and domains. Pfam is a member database of InterPro. The Pfam database is a large collection of protein families, each represented by multiple sequence alignments and hidden Markov models (HMMs)
![]()
EMBL-EBI
Example of protein database : HMMER
HMMER : Fast sensitive protein homology searches using profile hidden Markov models (HMMs) for querying against both sequence and HMM target databases.
![]()
EMBL-EBI
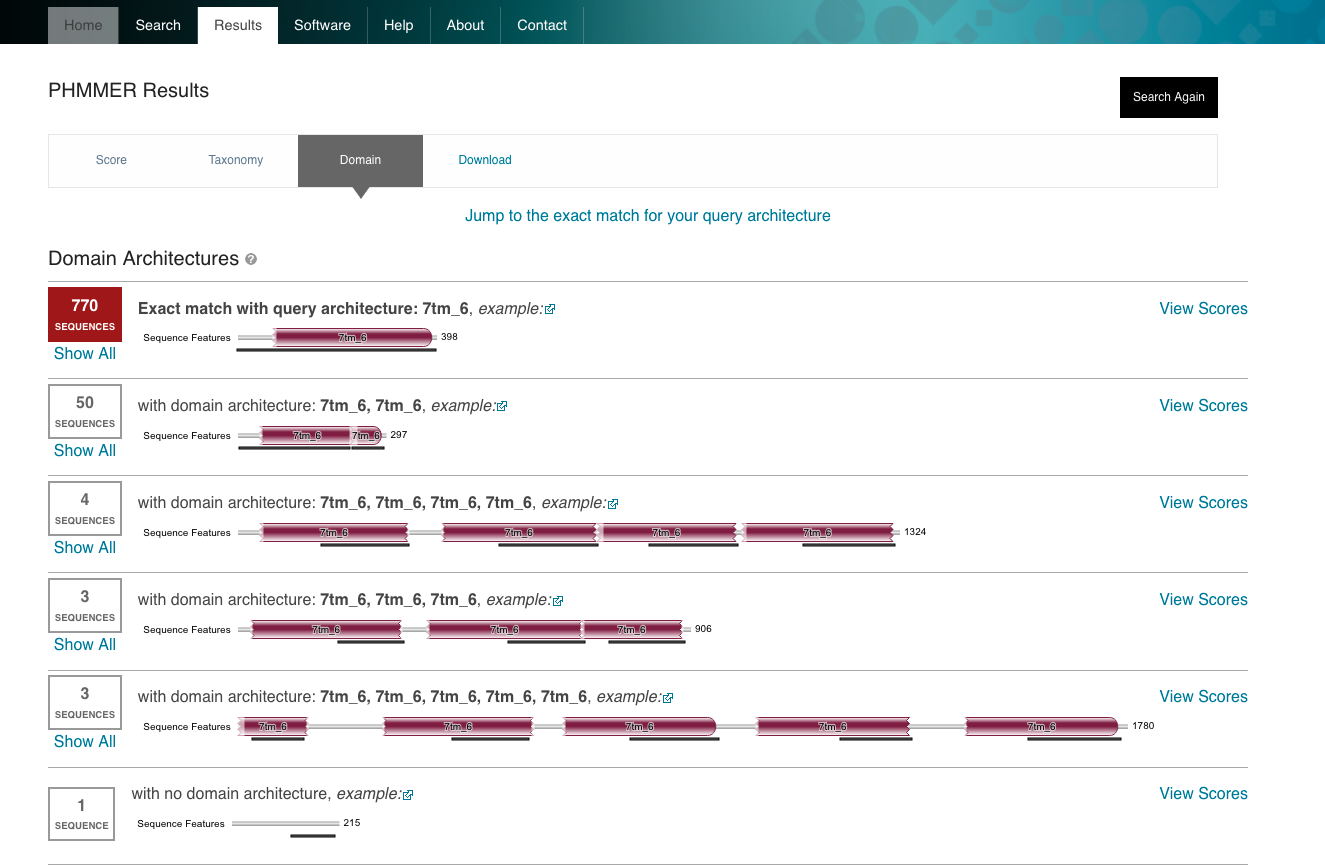
Example of protein database : HMMER
HMMER
![]()
NCBI
Example of DNA & RNA database : Nucleotide Database
Nucleotide Database : A collection of nucleotide sequences from several sources, including :
- GenBank : an annotated collection of all publicly available DNA sequences
- RefSeq : a comprehensive, integrated, non-redundant, well-annotated set of reference sequences including genomic, transcript, and protein.
- The Third Party Annotation (TPA) database : a database designed to capture experimental or inferential results that support submitter-provided annotation for, or assembly of, sequence data that the submitter did not directly determine but derived from GenBank primary data.
- Protein Database : a collection of sequences from several sources, including translations from annotated coding regions in GenBank, RefSeq and TPA, as well as records from SwissProt, PIR, PRF, and PDB.
Searching the Nucleotide Database will yield available results from each of its component databases.
NCBI
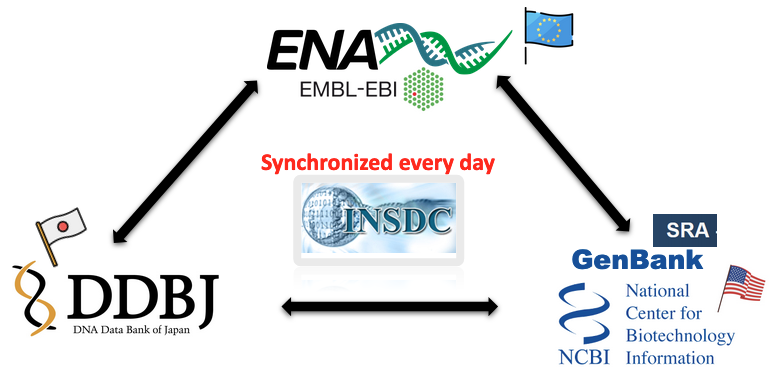
Example of DNA & RNA database : GenBank
GenBank : an annotated collection of all publicly available DNA sequences, is part of the International Nucleotide Sequence Database Collaboration , which comprises :
- the DNA DataBank of Japan (DDBJ),
- the European Molecular Biology Laboratory (EMBL),
- GenBank at NCBI.
GenBank consists of several divisions, most of which can be accessed through the Nucleotide database.
![]()
NCBI
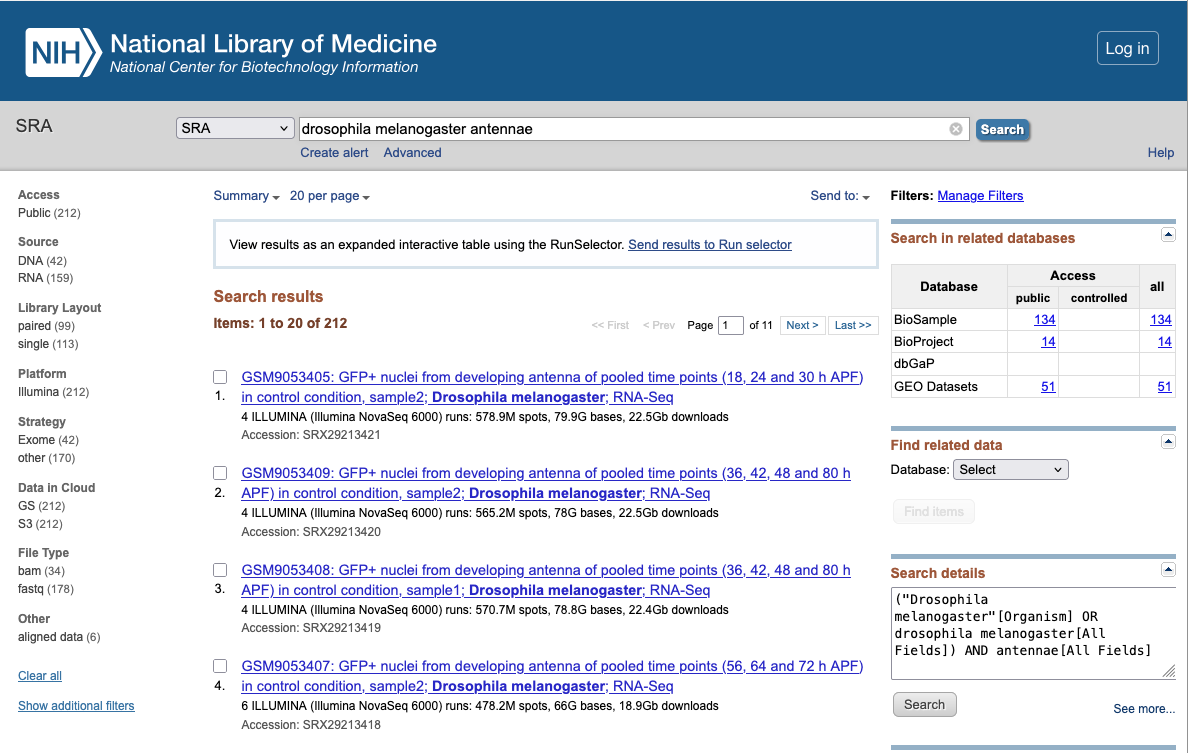
Example of DNA & RNA database : SRA (Sequence Read Archive)
SRA (Sequence Read Archive) : stores sequencing data from the next generation of sequencing platforms including Roche 454 GS System®, Illumina Genome Analyzer®, Life Technologies AB SOLiD System®, Helicos Biosciences Heliscope®, Complete Genomics®, and Pacific Biosciences SMRT®.
![]()
NCBI
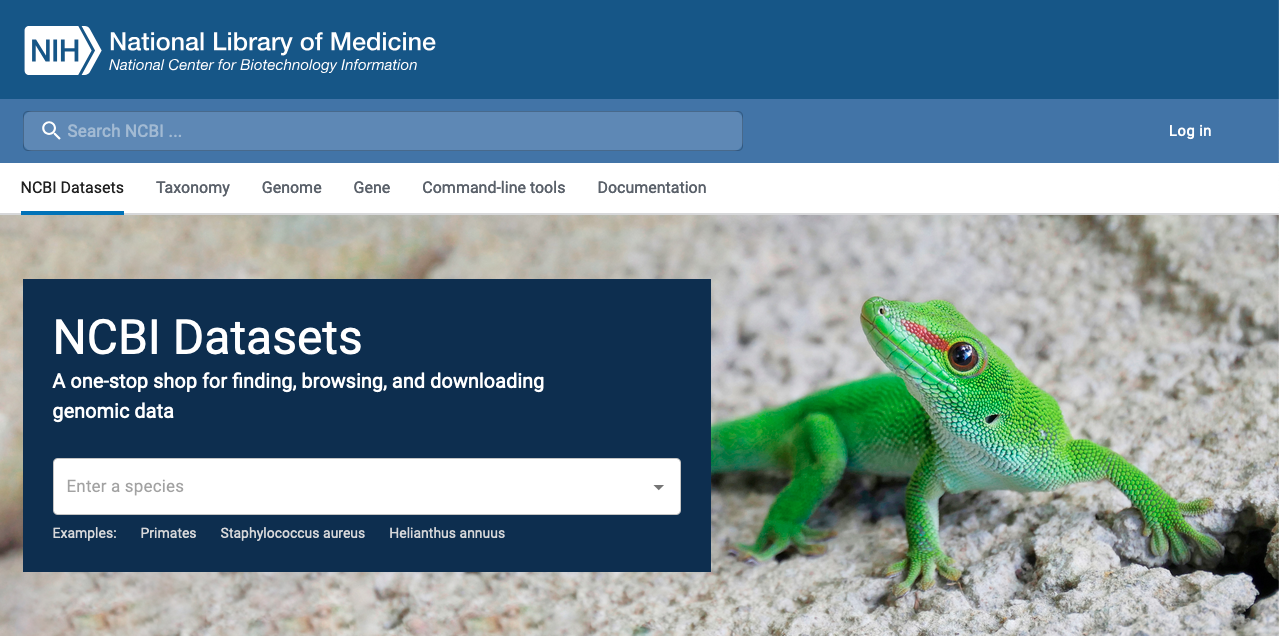
Example of DNA & RNA database : Genome
Genome : Contains sequence and map data from the whole genomes of over 1000 organisms.
![]()
NCBI
Example of DNA & RNA database : Genome
Genome
![]()
NCBI
Example of DNA & RNA AND protein database : RefSeq
RefSeq : A comprehensive, integrated, non-redundant, well-annotated set of reference sequences including genomic, transcript, and protein.
![]()
NCBI
Example of DNA & RNA AND protein database : RefSeq
RefSeq
![]()
NCBI
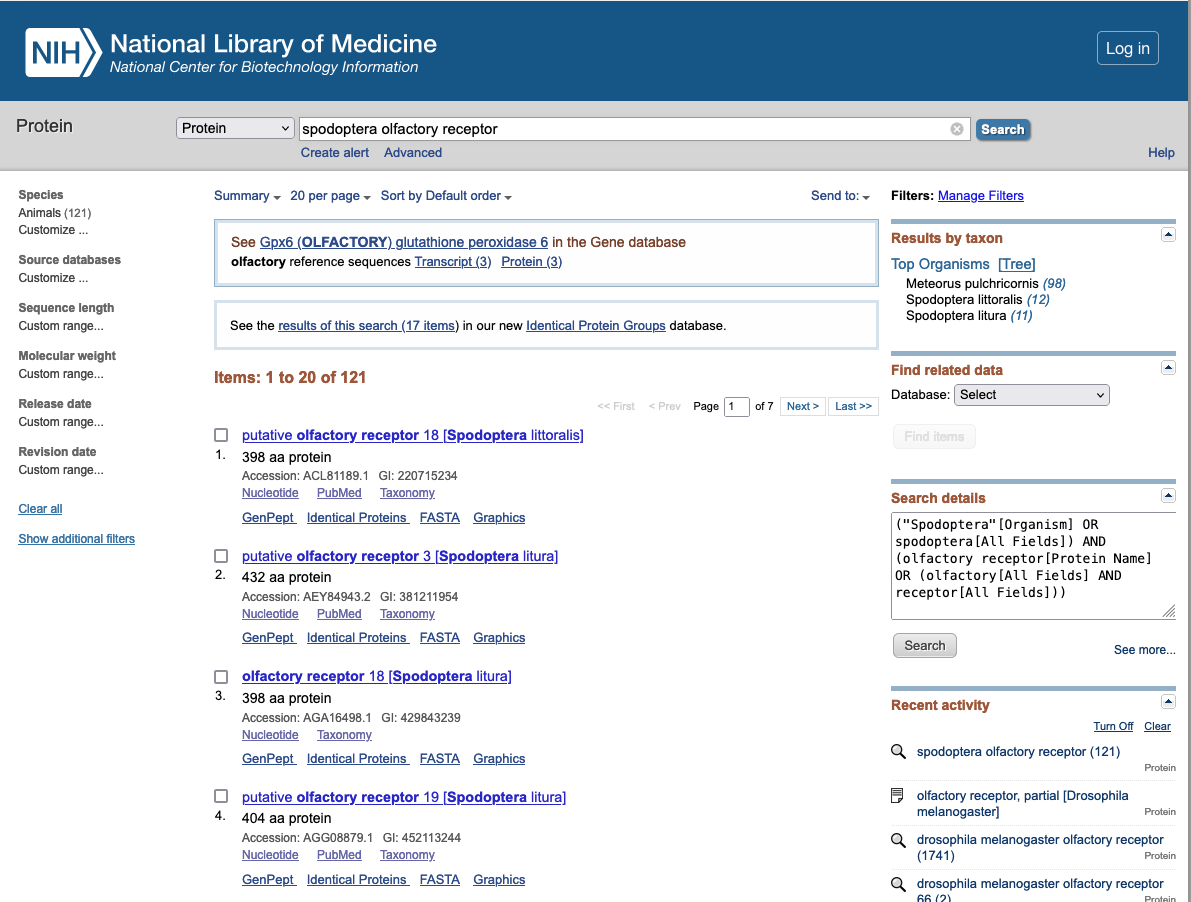
Example of protein database : Protein Database
Protein Database : includes protein sequence records from a variety of sources, including GenPept, RefSeq, Swiss-Prot, PIR, PRF, and PDB.
![]()
NCBI
Example of publication database : PubMed
PubMed : collection of biomedical books that can be searched directly or from linked data in other NCBI databases
![]()
NCBI
Example of publication database : PubMed
PubMed
![]()
NCBI
![]()
NCBI
![]()
NCBI
![]()
NCBI
![]()
Databases of specific genomes
Non-sequence-centric databases
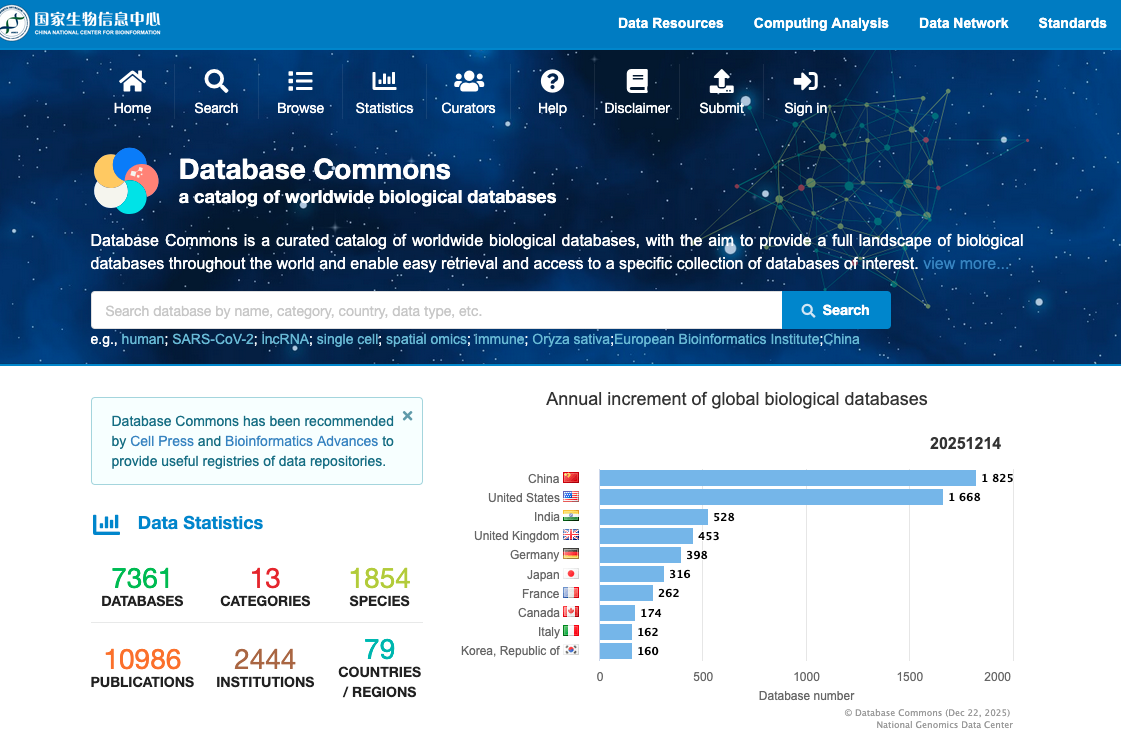
A catalog of worldwide biological databases
Database Commons is a curated catalog of worldwide biological databases, with the aim to provide a full landscape of biological databases throughout the world and enable easy retrieval and access to a specific collection of databases of interest
![]()
Public banks installed on the IFB cluster
Banks can be available in different versions and in different index format: fasta, gff, blast, bwa, bowtie, bowtie2, start, diamond, picard,
ls /shared/bank/
COG danio_rerio krona oryza_nivara saccharomyces_cerevisiae
MMSEQS_DB data.galaxyproject.org lachancea_kluyveri oryza_punctata saccharum_spontaneum
PFAM daucus_carota malus_domestica oryza_rufipogon salmonella_typhimurium
README.txt dbcan mgnify oryza_sativa_aus schizosaccharomyces_pombe
_scripts dram mmdb oryza_sativa_basmati software_specific
accession2taxid drosophila_melanogaster motus oryza_sativa_indica solanum_lycopersicum
alphafold2 eggnog-mapper mus_musculus oryza_sativa_japonica sorghum_bicolor
amrfinder elaeis_guineensis musa_acuminata oryza_sativa_nerica squeezemeta
anas_platyrhynchos emapperdb ncbi_taxonomy pdb sus_scrofa
arabidopsis_lyrata equus_caballus neofusicoccum_parvum pfam trypanosoma_brucei
arabidopsis_thaliana escherichia_coli nicotiana_tabacum phalaenopsis_aphrodite uniprot
astyanax_mexicanus felis_catus nr phalaenopsis_equestris uniprot_swissprot
bakta frogs nt phoenix_dactylifera uniref50
bos_taurus galaxy_test-data ophrys_sphegodes phyloflash uniref90
bowtie-index genbank oryctolagus_cuniculus plasmidfinder vanilla_planifolia
candida_glabrata greengenes oryza_barthii populus_trichocarpa vitis_vinifera
canis_lupus_familiaris gtdbtk oryza_brachyantha prunus_persica xenopus_laevis
capra_hircus hhsuite oryza_glaberrima rattus_norvegicus zea_mays
checkm homo_sapiens oryza_glumipatula refseq zymoseptoria_tritici
clostridium_perfringens kraken oryza_longistaminata rosa_chinensis
cocos_nucifera krakenuniq oryza_meridionalis rsat_organism
ls /shared/bank/uniprot_swissprot/
current uniprot_swissprot_2018-10-10 uniprot_swissprot_2020_03
ls /shared/bank/uniprot_swissprot/current/
blast diamond fasta flat listingv1.blast mapping mmseqs
Public banks installed on the IFB cluster
Banks can be available in different versions and in different index format: fasta, gff, blast, bwa, bowtie, bowtie2, start, diamond, picard,
The banks are organized first per species or bank names, then per index:
homo_sapiens/
├── GRCh38
│ ├── bwa
│ │ ├── Homo_sapiens.GRCh38.dna.primary_assembly.fa -> ../fasta/Homo_sapiens.GRCh38.dna.primary_assembly.fa
├── Homo_sapiens.GRCh38.dna.primary_assembly.fa.amb
├── Homo_sapiens.GRCh38.dna.primary_assembly.fa.ann
├── Homo_sapiens.GRCh38.dna.primary_assembly.fa.bwt
├── Homo_sapiens.GRCh38.dna.primary_assembly.fa.pac
└── Homo_sapiens.GRCh38.dna.primary_assembly.fa.sa
│ │ └── ...
│ ├── fasta
│ │ ├── Homo_sapiens.GRCh38.dna.primary_assembly.fa
├── Homo_sapiens.GRCh38.dna.primary_assembly.fa.fai
├── Homo_sapiens.GRCh38.dna.primary_assembly.fa.readme
├── Homo_sapiens.GRCh38.dna.toplevel.fa
├── Homo_sapiens.GRCh38.dna.toplevel.fa.fai
└── Homo_sapiens.GRCh38.dna.toplevel.fa.readme
│ │ └── ...
│ ├── gff3
│ │ ├── Homo_sapiens.GRCh38.94.gff3
│ │ └── Homo_sapiens.GRCh38.94.gff3.readme
│ └── star
│ ├── Genome
│ ├── Homo_sapiens.GRCh38.dna.primary_assembly.fa -> ../fasta/Homo_sapiens.GRCh38.dna.primary_assembly.fa
│ └── ...
└── hg19
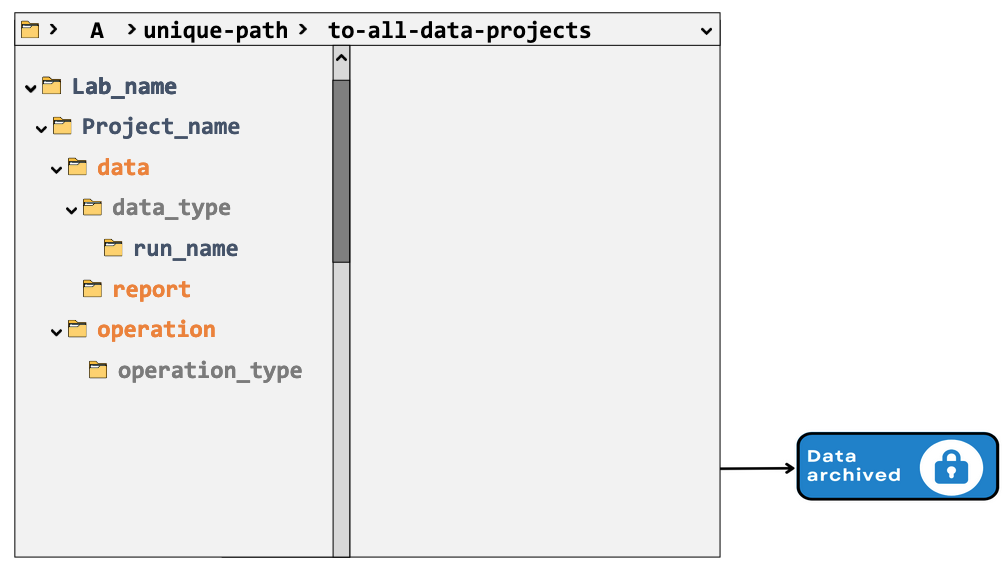
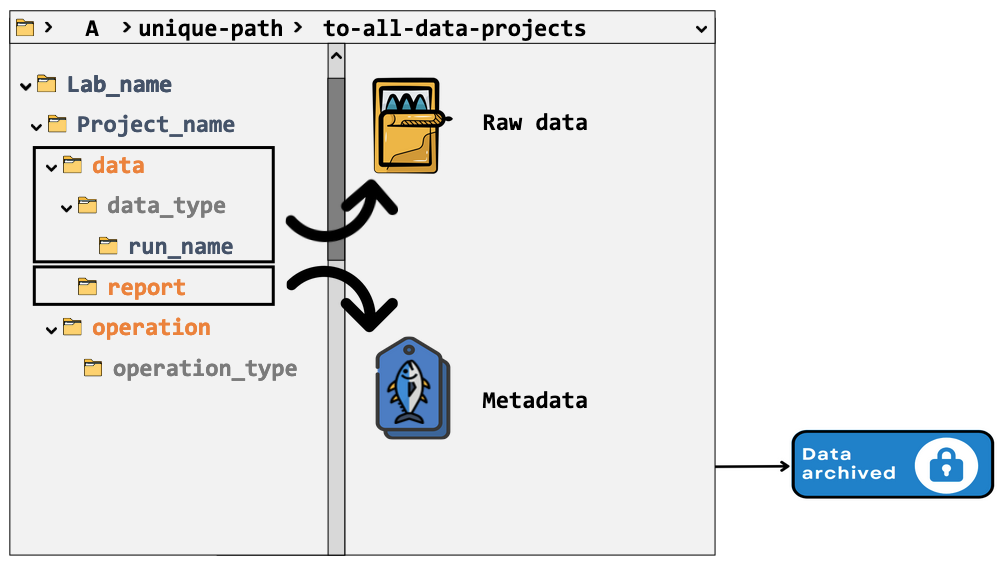
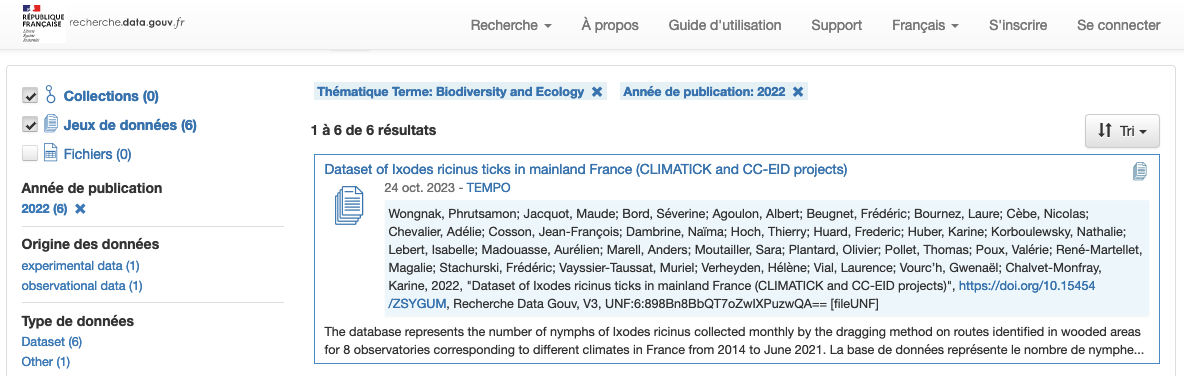
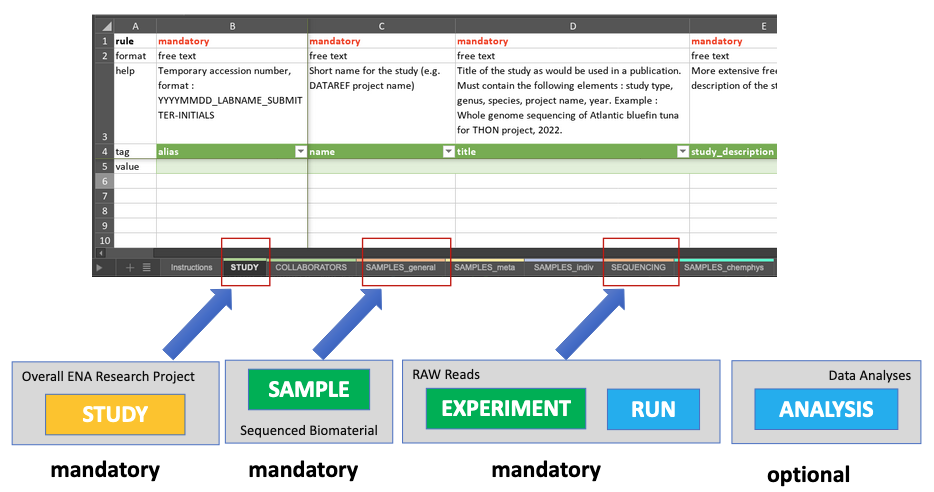
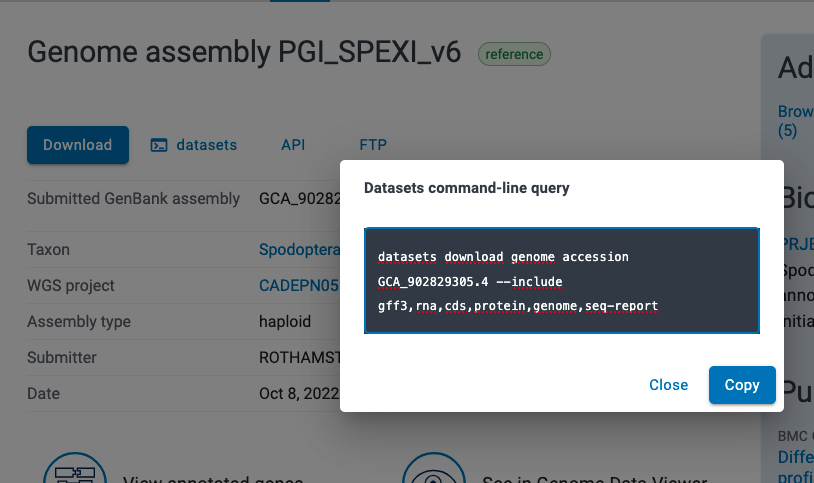
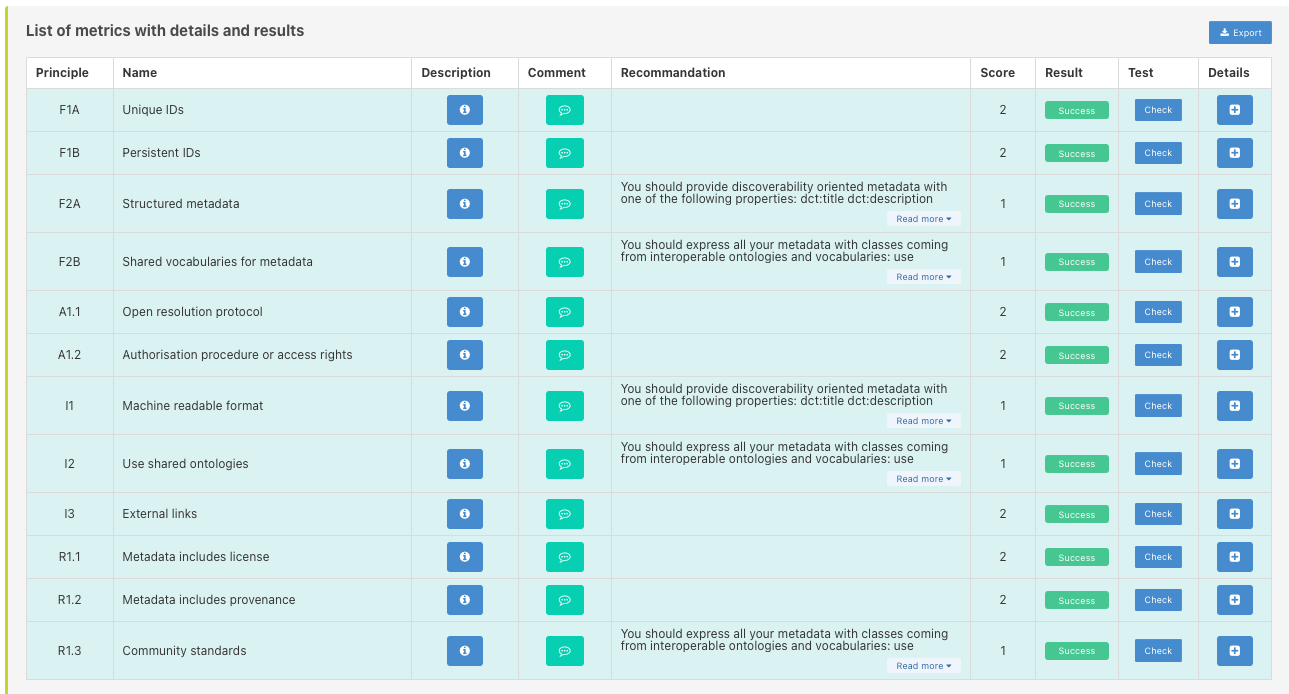
Data submission at ENA
Practical use of EBI-ENA data submission service
Let’s consider we want to publish our sequencing data within the European Nucleotide Archive (ENA) databank managed by the European Bioinformatics Institute (EBI), Hinxton, UK.
You have to:
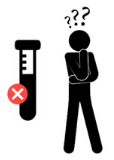
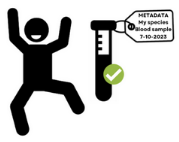
- Understand the metadata model to figure out what mandatory information you have to provide
- Collect all that information and setup document(s) ready for submission
- Submit metadata along with data using EBI tools
About ontologies
Ontologies are set of terms (along with unique IDs and description), each of them covering a particular scientific field and validated by scientific communities.
Ontologies are interoperable, logically well-defined, machine readable controlled vocabularies.
A few examples:
- The Gene Ontology (GO) 1 aims to maintain and develop controlled vocabulary of gene and gene product attributes
- EDAM 2 is an ontology of well established, familiar concepts that are prevalent within bioinformatics, including types of data and data identifiers, data formats, operations and topics.
- ENVO 3 is an ontology which represents knowledge about environments, environmental processes, ecosystems, habitats, and related entities
- and so on: more ontologies 4
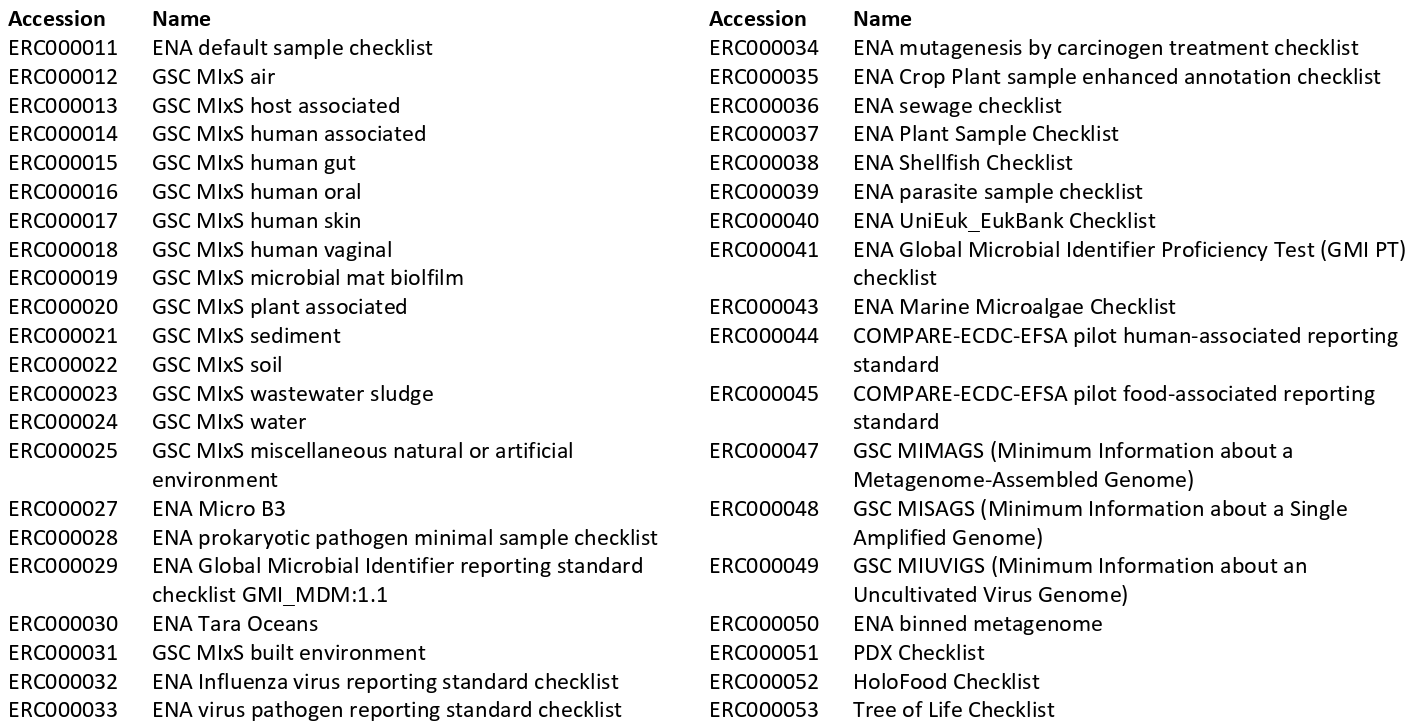
ENA checklists
As of November 2023, there are about 40 ENA sample checklists:
![]()
Do not invent your terms to describe your metadata. Instead, use above one!
ENA submission
- Time-consuming
- Difficult to handle
- Several tools available
ENA submission done
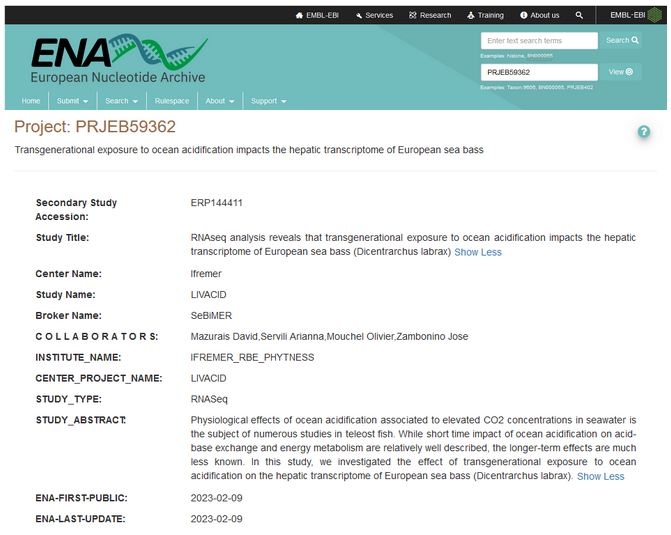
Example of a catalog entry programmatically submitted to ENA:
![]()
Metadata related to ENA Study object